分子轨道理论
原子轨道和分子轨道都是波函数。分子轨道是原子轨道交叠的结果,也就是原子轨道波函数相互干涉的结果。
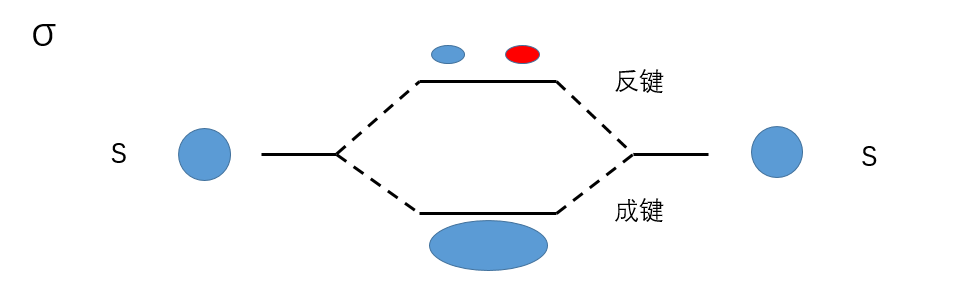
对于成键轨道σ1s,是两个原子轨道1sa和1sb相长干涉的结果:
可以发现相长干涉导致增加了项,导致原子间概率密度增加,能量降低,成键轨道能量降低。
对于反键轨道σ1s*,是两个原子轨道1sa和1sb相消干涉的结果:
可以发现相消干涉导致减少了项,导致原子间概率密度减少,能量增加,成键轨道能量增加。
原子轨道分布图描述的是解薛定谔方程得到的波函数ψ的值.函数值可以为正,也可以为负.形成共价键时要求同号重叠,即对称性匹配原则.
分析两个原子间成键数:成键数=0.5*(成键轨道电子数-反键轨道电子数)
2px和2py形成pi轨道,它们只是坐标不同,形成的成键和反键轨道能量相同,称为简并轨道。
O2中π2px*和π2py*上各有一个电子,称为二价自由基,凡是有未成对电子都称为自由基,要考虑其磁性。
价电子成键理论
两个未配对电子导致的成键
杂化理论
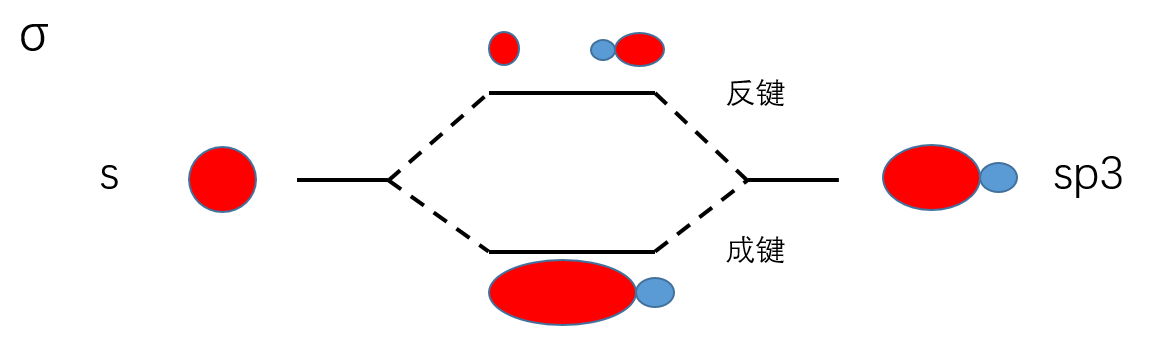
s和p轨道杂化产生的spx轨道能量高于s轨道,低于p轨道,方便成键和形成单电子。杂化所需要的能量由成键时释放的能量提供。
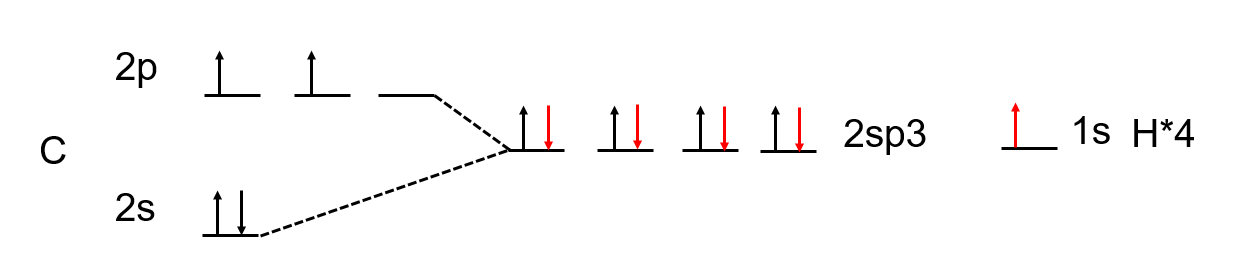
sp3杂化中小的一端是反键,大的一端是成键。
轨道形状
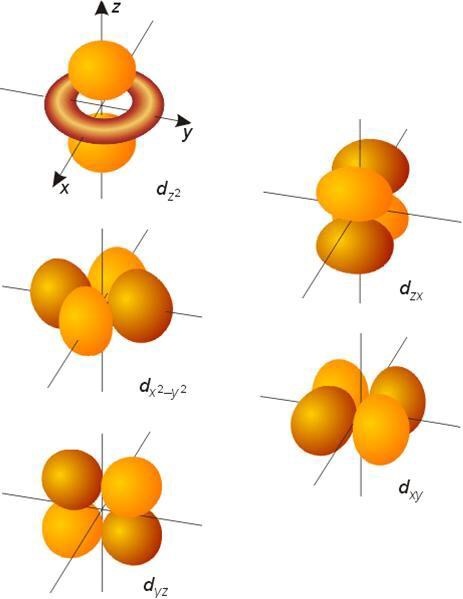
d轨道
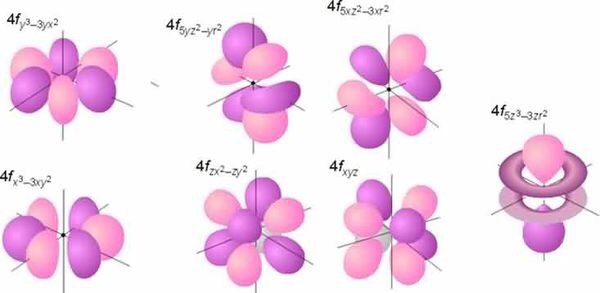
f轨道
轨道成键形状
下面列举各种相同或不同轨道成键例子
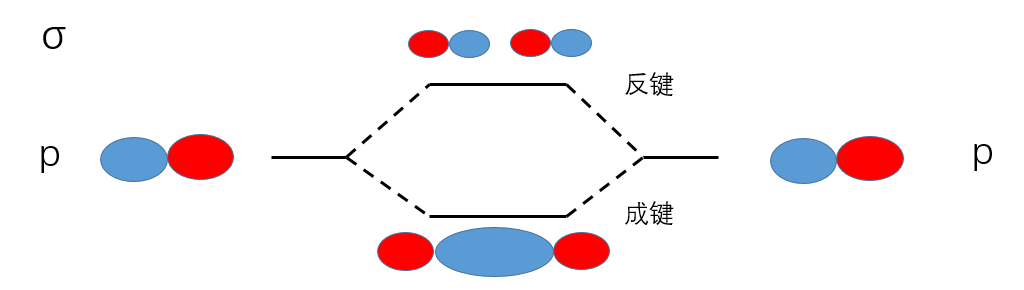
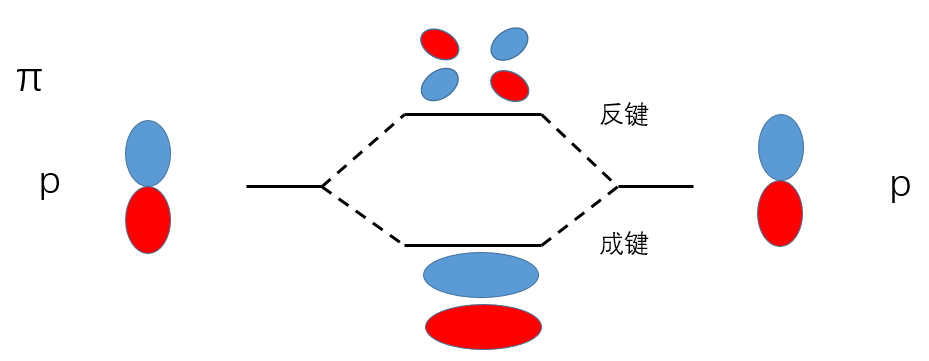
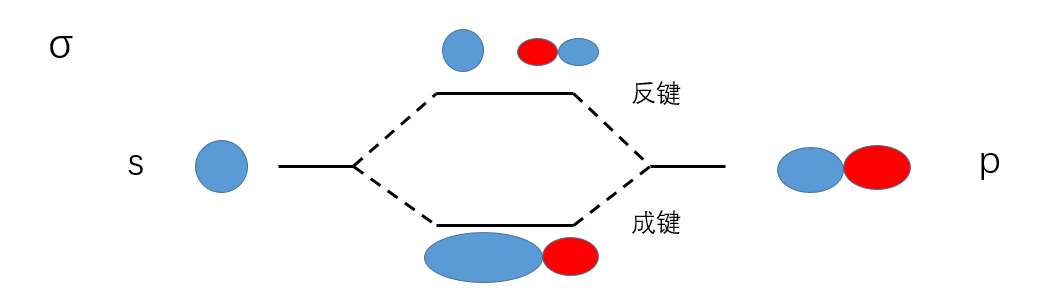
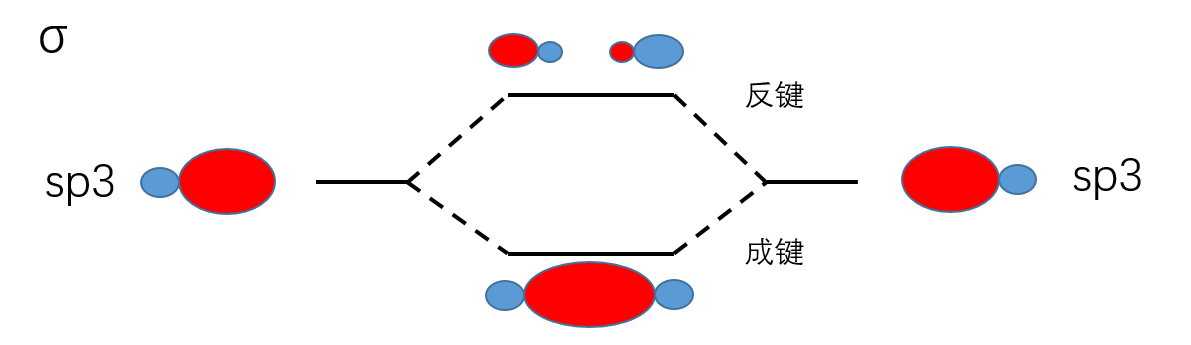
周期性体系
周期性体系分子轨道就变成了能带的概念。能带计算首先计算单点能,保留CHGCAR。
吸附具体过程
但如果只是局部的金属的模型仍然可以用金属原子轨道来讨论
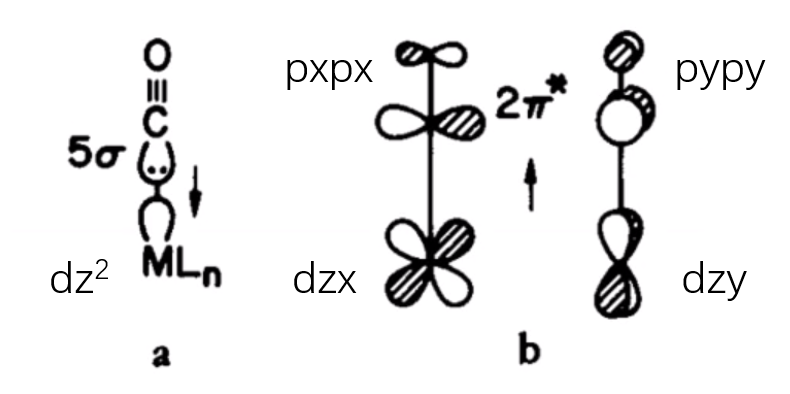
距离CO和Ni表面的成键方式,CO的σ轨道与Ni的dz2轨道头碰头结合,CO的2π*反键轨道与Ni的dzx,dzy以肩并肩方式成键。
活化:活化为吸附后,金属原子的d轨道电子反馈给吸附分子2π*反键轨道电子,因此吸附分子本身的键(如CO中C与O的键)变弱,键长变长。具体表现在DOS图中2π*反键轨道在成键后部分下移到费米能级之下,得到部分电子。因此反键轨道部分移到费米能级下面是活化的证据。
通过DOS可以看出活化过程,而配合ssNBO和COHP可以进一步分析键的成键和键强的变化。
而电荷差分和自旋密度可以分析电子的分布状况。
DOS计算模型准备
切好晶面后添加真空层,固定金属下面几层以准备表面优化,把吸附分子放在真空层(15A)中间,避免吸附分子吸附在上表面或者下表面。这样可以对齐吸附分子和表面的费米能级,方便电子结构分析。对比吸附前和吸附后的DOS图
一般表面计算参数都设置(LREAL = Auto),ISYM=0去掉对称性
DFT计算时吸附分子之间相距10A,相互作用可以忽略不计
DOS中吸附和金属轨道的重叠可以代表成键的贡献
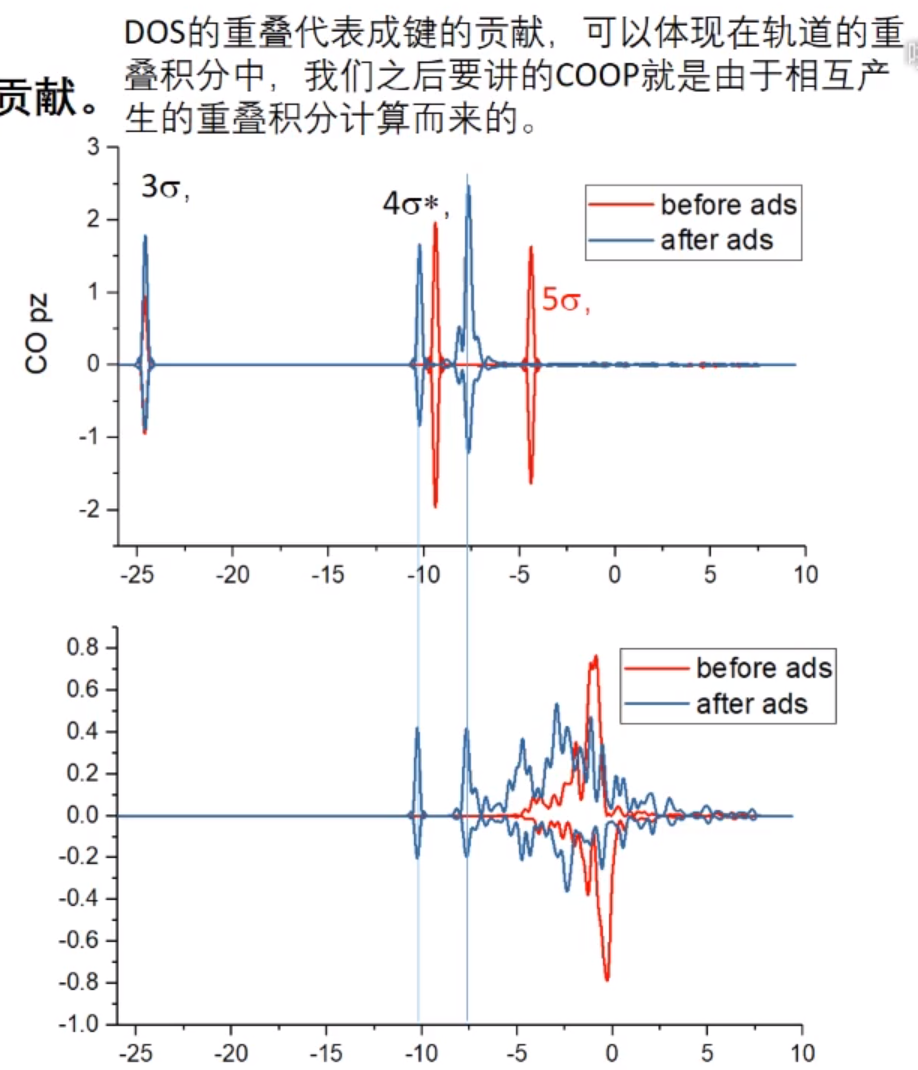
查看不同的轨道对总轨道的贡献,画出单独的吸附质分子的轨道图,分清sigma键和pi键对应的轨道。再画出DOS图,查看是s、px、py、pz哪个轨道给sigma或pi轨道作贡献。
单个吸附分子的DOS和轨道图计算
首先单独优化吸附分子,需要输出WAVECAR
然后查看EIGENVAL文件(cat EIGENVAL),明确电子占据情况
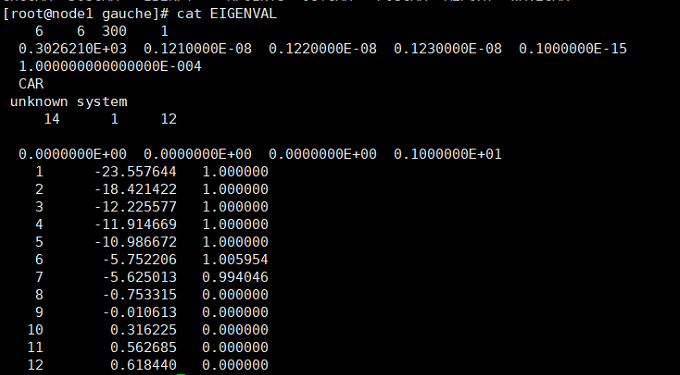
最左边是能带的编号(NBAND),第二列是能带的能量(eV),第三列是电子占据数
可以使用vaspkit511的功能来画出轨道图
脚本
for i in $(seq 1 7)
do echo $(i)
echo -e "51\n511\n1\n${i}\n" | vaspkit
done
分子轨道形成与DOS图关系
轨道形成三原则:对称性一致,能量相近,轨道最大重叠。可以根据这三个原则对DOS图分析吸附的具体作用情况。
对称性一致:体现在DOS图中吸附分子是sigma轨道吸附后发生明显变化还是pi轨道发生明显变化;
能量相近:体现在吸附分子与吸附基底能级的位置
轨道重叠:体现在形变后吸附分子和吸附基底的能级轨道是不是位置贴近。
化学键的进一步研究:COOP,COHP
COOP
就是DOS图乘以重叠布局权重,重叠布局只可以在两个原子之间,因此COOP也只可以在两个原子之间。
重叠布局:分子轨道波函数为原子轨道的线性组合:
电子分布的概率是归一化的:
其从是两个原子波函数之间的重叠积分。是相互作用有关的量,称为重叠布局。如果同号,则为正值(成键),如果反号,则为负值(反键)。
因此乘以重叠布局权重后可以判断成键和反键,重叠程度大小情况。COOP费米能级以下的积分值可以反应键强。对比吸附前后的COOP可以分析键强的变化。
d带中心
配位数增加可以使d带更宽,d带中心下降,降低吸附能。
当晶格中分子之间距离边长,d带宽度变窄,吸附能增加。
DFT分析金属稳定性
可以分析空位形成能来分析稳定性(DOI: 10.1021/jacs.8b08836)