1 DOS计算
1.1 单个气体分子的轨道图计算
首先单独优化吸附分子,需要输出WAVECAR
然后查看EIGENVAL文件(cat EIGENVAL),明确电子占据情况
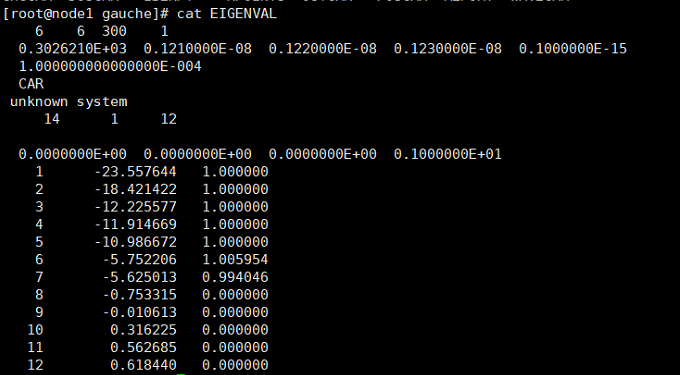
最左边是能带的编号(NBAND),第二列是能带的能量(eV),第三列是电子占据数
可以使用vaspkit511的功能来画出轨道图
使用该功能后会输出RWAV_B000x_K000x.vasp文件,用vesta打开可以看
脚本
for i in $(seq 1 7)(这里要写出输出的轨道数,从EIGENVAL获得)
do echo $(i)
echo -e "51\n511\n1\n${i}\n" | vaspkit (-e 是开启识别执行转移字符功能,从而其中的\n是回车)
done
或者使用脚本
#!/usr/bin/env bash
for i in $(seq 1 $1 )
do
echo $i
echo -e "51\n511\n1\n${i}\n" | vaspkit
done
然后命名为orbital,使用orbital+需要输出的轨道数,直接得到
1.2 DOS
LORBIT = 11
NEDOS = 2000
IBRION = -1(原子不动)
1.2.1 单个气体分子111计算
K点会使用111
INCAR文件需要注意的是
ISMEAR = 0(不用-5)
SIGMA = 0.05
1.2.2 slab吸附模型的DOS
可以做一个吸附模型,一个吸附分子不吸附,在空中的模型,方便费米能级的对比。
可以优化的时候直接放LORBIT=11,只要晶格常数不变,就可以直接优化。
ISYM=0 对称性为0,表面计算引入对称性可能导致表面不能充分弛豫。
IDIPOL=3可以去除偶极,但是对计算能量和电子结构时影响不大,可以不添加。计算功函时要添加。